Genetic variation in parasite crosses
About the crosses
The malaria parasite Plasmodium falciparum is a tiny, single-celled organism, uniquely adapted to living inside humans and mosquitoes. It is, however, a eukaryote, which means it shares more in common with animals and plants than it does with bacteria or viruses. For example, malaria parasites reproduce sexually. When they are taken up by a mosquito bite, they differentiate into male and female gametes, which fuse to form a zygote. As a zygote, the parasite is diploid, meaning it has two copies of each of the chromosomes that make up its genome, like the cells in our bodies. However, it doesn’t stay like that for long, undergoing meiosis shortly afterwards. During meiosis the cell divides, transmitting only a single genome copy to each daughter. The rest of the parasite’s life cycle is spent as a haploid. If you’ve ever heard the joke about how man is but a brief stage in the life cycle of a sperm, for malaria parasites that is not far from the truth, at least from a genomic point of view.
Because malaria parasites have sex, they can be crossed. A parasite cross is similar in principle to a cross with any other organism. Choose two “parents” that differ in some trait of interest (e.g., resistance to an anti-malarial drug), give them the opportunity to mate, collect their “children”, and study how the trait has been inherited. In practice it’s not quite so simple, because you cannot actually cross two individual parasites. Rather you have to cross two parasite clones, meaning that each “parent” is actually many thousands of parasites which have been cultured to be genetically identical. However, Walliker et al. (1987) showed that it can be done, performing the first cross between clones 3D7 and HB3. Wellems et al. (1991) performed a second cross between clones HB3 and Dd2, which they used to discover the gene responsible for chloroquine resistance. Hayton et al. (2008) performed a third cross between clones 7G8 and GB4, discovering a gene involved in parasite invasion of human red blood cells, now a promising vaccine candidate.
Data release and web application
The MalariaGEN P. falciparum genetic crosses project was set up in 2012 with the goal of deep sequencing the parents and progeny of these three crosses. The project has given us some unique insights into the unusual world of P. falciparum genome biology, and it’s been a great privilege to take part in the analysis. We recently made a public data release from the project, which includes SNP and INDEL calls from two different methods of variant discovery, as well as the underlying sequence data itself. We also created a web application to help explore the data (see the screen cast below for a brief tour), and posted a preprint on biorxiv describing the data and what it can tell us about how parasites evolve to face new challenges, such as pressure from anti-malarial drugs.
Notes on variant calling
The crosses are a great proving ground for variant calling methods. Each cross comprises two parents and up to 35 progeny clones. We sequence at the haploid merozoite life cycle stage, so there is no heterozygosity to worry about, calling a genotype means calling a single allele. And for the progeny, we know their genotype must be inherited from one parent or the other. True de novo mutations are relatively rare, so any variant where an allele is called in one or more progeny clone that is not found in either parent is probably a “Mendelian” error. Furthermore, for some progeny there are multiple biological replicates, which means we sequenced the same clone several times. These replicates should be identical, so any disagreement between them is also probably an error. These features of the dataset provide a means to empirically calibrate and evaluate the outputs from a variant caller.
To call SNPs and small INDELs, we used two methods. The first was based on alignment of sequence reads to the 3D7 reference genome using BWA, followed by GATK (BQSR, INDEL realignment, UnifiedGenotyper, VQSR). The second used Cortex via the independent workflow, which involves a De Bruijn graph assembly for each clone, followed by mapping of assembled variants back to the 3D7 reference genome to obtain coordinates. These two methods work in very different ways, so we wanted to learn about their relative strengths and weaknesses.
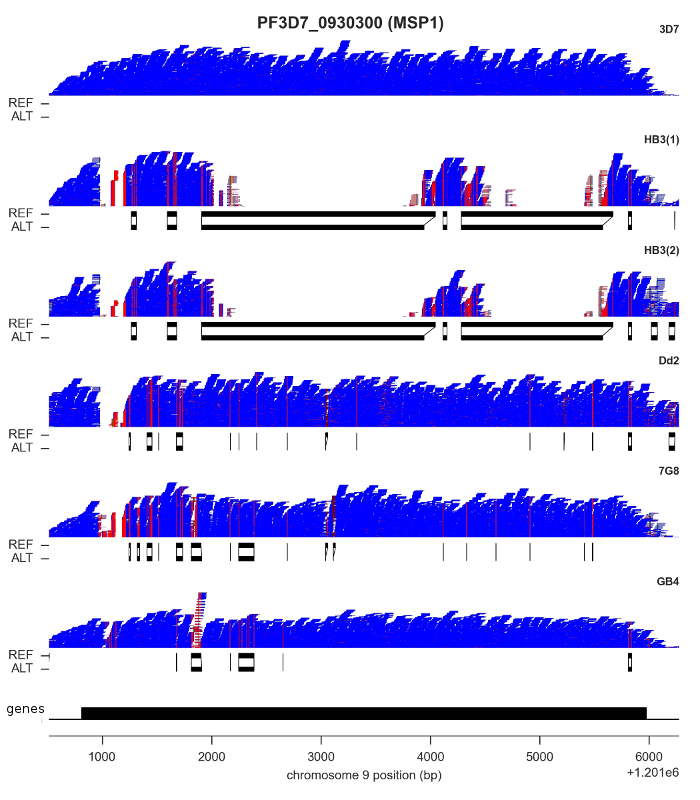
For both SNPs and INDELs, we found both methods could be calibrated to produce highly accurate results: low rates of Mendelian error, and almost perfect concordance between replicates. However, although SNP density is relatively low throughout most of the core genome, there are a handful of genes where some clones have sequences that are highly diverged, with as little as ~60% identity with the reference genome. I’ve shown the most extreme example, MSP1, in the figure above. Here, clone HB3 diverges from the reference genome in two separate regions. You can see this from the read pileups, because mapping fails completely. If you didn’t know better, you might think there was a deletion here. Below the pileups, however, I’ve plotted the reference and alternate sequences from the “bubbles” assembled by Cortex. This gene has previously been capillary sequenced, so we know that Cortex’s assemblies are correct.
It is probably no coincidence that most of the genes where you find this kind of variation are expressed on the surface of the blood-stage parasite, and so are likely to be involved either in evading the human immune system or invading red blood cells. These are medically important processes, so being able to get a complete picture of parasite genetic variation is vital. We were very pleased to find Cortex was able to access this kind of variation, because it opens the possibility of high-throughput studies in parasites collected from the field, which is now being followed up in the Pf3k project.
Cortex is, however, by no means perfect. Currently it can only genotype biallelic variants. For INDELs especially, we found a substantial number of multiallelics in the HB3xDd2 and 7G8xGB4 crosses (based on results from GATK), so in those crosses Cortex was limited. Also, Cortex struggles in the non-coding regions of the P. falciparum genome. Non-coding regions are AT-rich, which means they tend to be underrepresented in the sequence data. To cope with lower coverage, Cortex needs to use a smaller K-mer size to achieve sufficient overlap between reads. However, non-coding regions are also low in sequence complexity, and need longer K-mers to create a unique assembly. The result is a lower rate of variant discovery and a higher rate of genotype missingness.
So in this study we found GATK and Cortex were complementary, compensating for each other’s weaknesses to a certain extent. For the analyses in the paper we combined the results from both methods into a single call set. Further details are available in the supplementary information associated with the preprint.
Further reading
- Walliker, D., Quakyi, I. A., Wellems, T. E., McCutchan, T. F., Szarfman, A., London, W. T., … Carter, R. (1987). Genetic analysis of the human malaria parasite Plasmodium falciparum. Science (New York, N.Y.), 236(4809), 1661–6.
- Wellems, T. E., Walker-Jonah, A., & Panton, L. J. (1991). Genetic mapping of the chloroquine-resistance locus on Plasmodium falciparum chromosome 7. Proceedings of the National Academy of Sciences of the United States of America, 88(8), 3382–6.
- Hayton, K., Gaur, D., Liu, A., Takahashi, J., Henschen, B., Singh, S., … Wellems, T. E. (2008). Erythrocyte binding protein PfRH5 polymorphisms determine species-specific pathways of Plasmodium falciparum invasion. Cell Host & Microbe, 4(1), 40–51.
- Miles, A., Iqbal, Z., Vauterin, P., Pearson, R., Campino, S., Theron, M., … Kwiatkowski, D. (2015). Genome variation and meiotic recombination in Plasmodium falciparum: insights from deep sequencing of genetic crosses. bioRxiv. Cold Spring Harbor Labs Journals.